单细胞转录组数据分析之Seurat标准流程
Yunshun Chen
Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Melbourne, Australiayuchen@wehi.edu.au
Jinming Cheng
Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Melbourne, Australiacheng.j@wehi.edu.au
September 26, 2021
Source:vignettes/scRNAseqWorkshop-Chinese.Rmd
scRNAseqWorkshop-Chinese.Rmd引言
单细胞RNA测序 (scRNA-seq)已经成为了被广泛使用的技术,能够让研究人员在单细胞水平测得基因表达图谱和研究分子生物学机制。 它提供了传统bulk RNA实验无法实现的细胞群体的生物学分辨率。.
这里, 我们提供了一套详细的工作流程,用于分析来自于人类乳腺组织单细胞RNA表达图谱(Pal et al. 2021)的10X单细胞RNA-seq数据。 这个细胞图谱探究了不同状态下人类乳腺组织的细胞异质性,包括正常, 癌前和癌变状态。
我们将会使用这套细胞图谱数据的一部分来展示如何对一个单细胞样本进行标准分析。 大部分的分析将会使用 Seurat (Satija et al. 2015) 包来完成。
初步准备
原始数据预处理
原始10X数据是BCL格式。 它们需要被软件工具提前处理比如 cellranger。
我们先用 cellranger 将BCL文件转换成FASTQ文件, 然后再将reads比对到人类基因组, 最后定量每个细胞中的每个基因的UMI counts。 这个工作流程使用的是 cellranger 的输出文件,并且所有分析过程都是在R环境中进行,。 我们没有包括 cellranger 运行的详细过程, 因为这不在此工作流程的范围之内。 关于运行 cellranger 的详细信息可以在这里获得。
下载read counts
在本次workshop中,我们会用到这个已发表的研究(Pal et al. 2021)中的一个样本。 该样本对应的病人ID如下:N1469, 其GEO登记号是GSM4909258。
我们先在当前工作目录下创建一个叫 Data 的文件夹。 然后我们在 Data 文件夹下面用病人的ID名创建一个子文件夹。
OneSample <- c("N1469")
out_dir <- file.path("Data", OneSample)
if (!dir.exists(out_dir)) dir.create(out_dir, recursive = TRUE)cellranger的输出结果包含三个数据文件:一个 mtx.gz 格式的count矩阵文件,一个 tsv.gz 格式的barcode信息文件,和一个 tsv.gz 格式的feature信息文件。 我们从GEO下载了这个样本的三个数据文件,并将它们放在子文件夹中。
GSM <- "https://ftp.ncbi.nlm.nih.gov/geo/samples/GSM4909nnn/"
url.matrix <- paste0(GSM,
"GSM4909258/suppl/GSM4909258_N-NF-Epi-matrix.mtx.gz")
url.barcodes <- gsub("matrix.mtx", "barcodes.tsv", url.matrix)
url.features <- "https://ftp.ncbi.nlm.nih.gov/geo/series/GSE161nnn/GSE161529/suppl/GSE161529_features.tsv.gz"
utils::download.file(url.matrix, destfile=paste0("Data/", OneSample, "/matrix.mtx.gz"), mode="wb")
utils::download.file(url.barcodes, destfile=paste0("Data/", OneSample, "/barcodes.tsv.gz"), mode="wb")
utils::download.file(url.features, destfile=paste0("Data/", OneSample, "/features.tsv.gz"), mode="wb") 标准分析
读入数据
我们加载了Seurat包,并且读取了病人N1469的10X数据. N1469.data对象是一个包含病人N1469的原始count数据的稀疏矩阵。 行是特征(基因),列是细胞。 默认情况下, 数据的列名是细胞barcode。
library(Seurat)
N1469.data <- Read10X(data.dir = "Data/N1469")
colnames(N1469.data) <- paste("N1469", colnames(N1469.data), sep="_")然后我们创建了一个名为N1469的Seurat对象。 在少于3个细胞中表达的基因被去除了。 细胞至少有200个基因被检测到才会被保留下来。
N1469 <- CreateSeuratObject(counts=N1469.data, project="N1469", min.cells=3, min.features=200)质量控制
质量控制是单细胞测序分析的基本步骤。 低质量的细胞和低表达的基因应该在分析之前去除。
两种常用的评估细胞质量的方法是看每个细胞的文库大小(library size)和表达的基因数目。 在使用 CreateSeuratObject()创建Seurat对象时,基因数目和总的分子数(library size) 会被自动计算。 另一种评估方法是看每个细胞中来自于线粒体基因的reads比例。 具有高线粒体reads含量的细胞更倾向于死亡,因此,它们应该在分析之前被去掉(Ilicic et al. 2016)。 这里,我们计算了来自线粒体基因的reads的百分比,并将其保存在Seurat对象的metadata中。 我们把“MT-”开头所有基因当作一个线粒体基因集。
N1469[["percent.mt"]] <- PercentageFeatureSet(N1469, pattern = "^MT-")一个Seurat对象的QC指标可以通过如下方式查看。
head(N1469@meta.data)## orig.ident nCount_RNA nFeature_RNA percent.mt
## N1469_AAACATACGGAGCA-1 N1469 7459 1870 2.65
## N1469_AAACATTGCCTCAC-1 N1469 5457 1393 1.89
## N1469_AAACATTGGTAGGG-1 N1469 7547 1964 2.09
## N1469_AAACCGTGCAGTTG-1 N1469 765 328 38.17
## N1469_AAACCGTGCATGAC-1 N1469 3908 1294 5.12
## N1469_AAACGCACTCAGGT-1 N1469 7218 2102 2.13可以通过画散点图来可视化一些QC指标。
plot1 <- FeatureScatter(N1469, feature1 = "nCount_RNA", feature2 = "percent.mt", plot.cor=FALSE)
plot2 <- FeatureScatter(N1469, feature1 = "nCount_RNA", feature2 = "nFeature_RNA", plot.cor=FALSE)
plot1 + plot2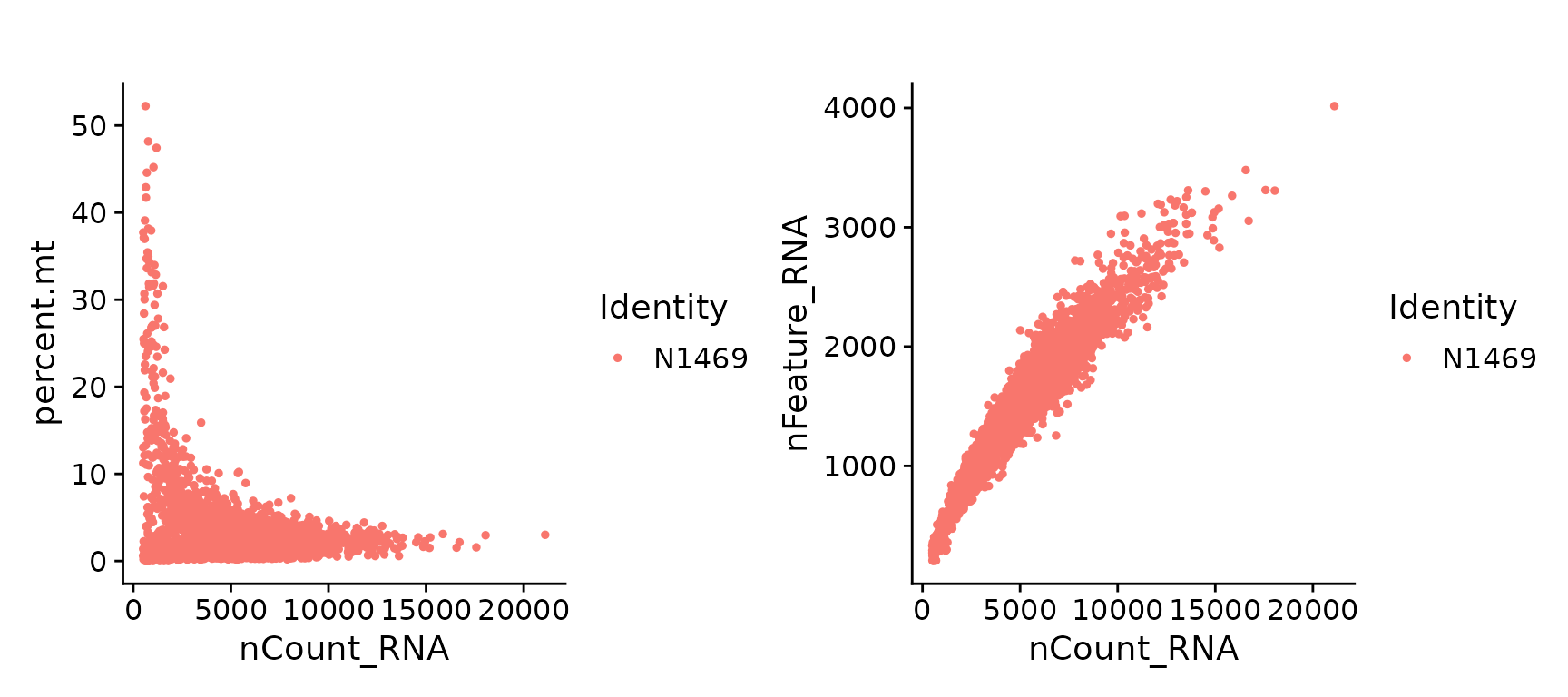
QC指标的散点图
对这套特定数据而言,我们过滤掉了那些唯一特征(基因)小于500并且线粒体counts比例大于20% 的细胞。
N1469 <- subset(N1469, subset = nFeature_RNA > 500 & percent.mt < 20)标准化
在过滤细胞之后,接下来的一步是标准化。 标准化在去除细胞特异偏好上很有用。
这里,我们使用了Seurat中默认的标准化方法,其先用每个细胞中的基因counts数除以总的counts数,再乘以一个缩放因子10000,最后对结果取对数。
N1469 <- NormalizeData(N1469)高度变化基因
单细胞RNA-seq数据经常被用来探究细胞群体之间的异质性。 为了减少下游分析的计算复杂度并且专注于真实的生物信号,在进行下游分析之前,经常会挑选一个高度变化基因(HVG)子集。 最常用的策略之一是选择那些在所有细胞中具有最高方差的基因。 高度变化基因的数目选择是相当随意的,任何在500到5000之间的数值都被认为是合理的。
对于这套数据,我们选择了最高度变化的1500个基因用于下游分析,比如PCA和UMAP可视化。
N1469 <- FindVariableFeatures(N1469, selection.method="vst", nfeatures=1500)均值-方差图可以被用来可视化变化最大的基因。
top50 <- head(VariableFeatures(N1469), 50)
plot1 <- VariableFeaturePlot(N1469)
plot2 <- LabelPoints(plot=plot1, points=top50, repel=TRUE)
plot2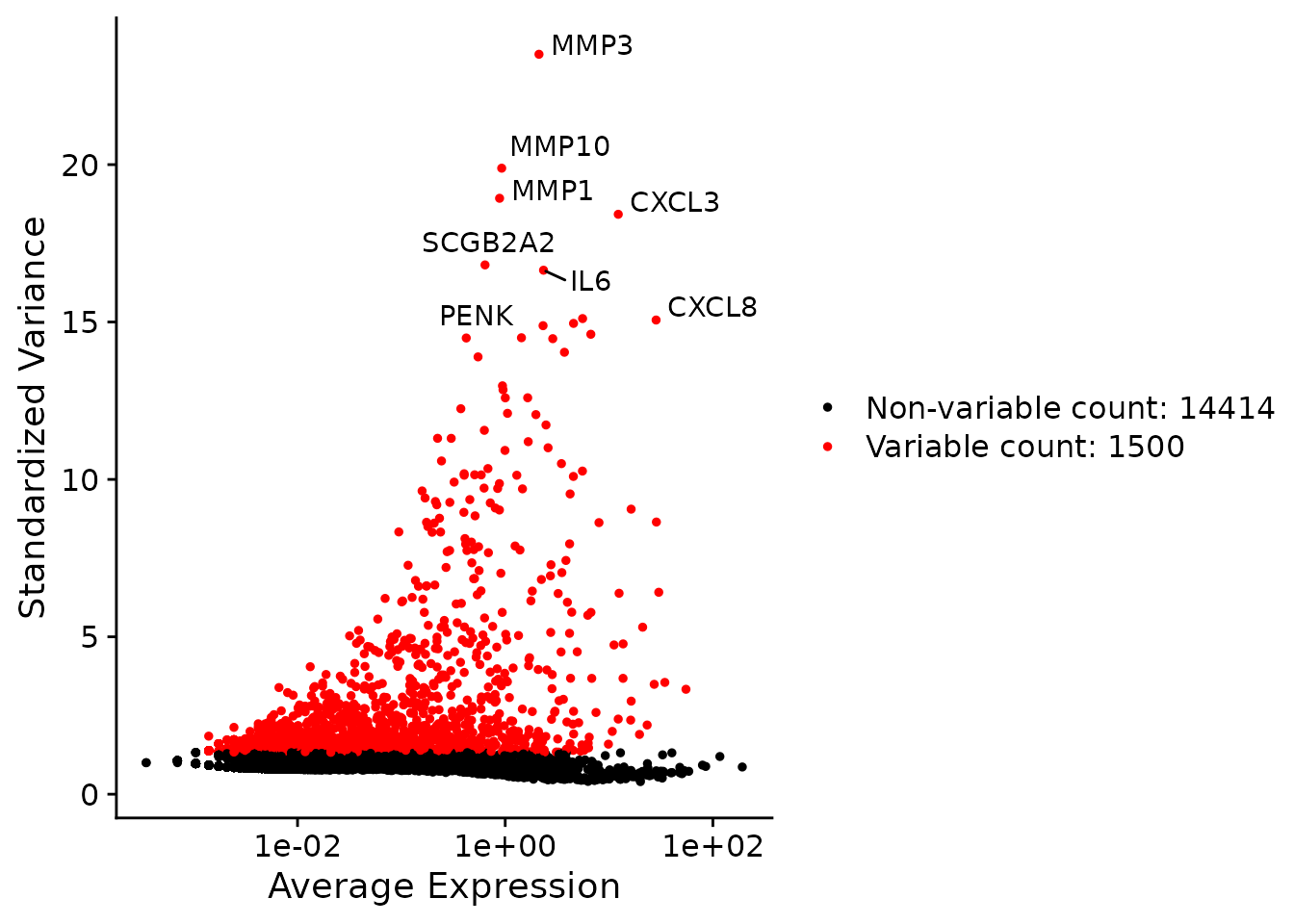
均值-方差图,最高度变化的1500个基因用红色高亮,最高度变化的50个基因被标记出来
在进行降维之前,我们使用了线性转化来缩放(scale)数据。 这种数据缩放是通过ScaleData()函数来完成,其标准化了每个基因的表达,使表达量在所有细胞中的均值为0,标准差为1。 这一步赋予了用于下游分析的基因相同的权重,所以高表达的基因不会占据主导。
默认情况下,缩放过程只会被应用于之前找到的1500个高度变化基因,因为这些基因被用在了下游分析中。
N1469 <- ScaleData(N1469)降维
降维是单细胞分析中的一个基本步骤。 它在一个相对较低的维度概括了上千个基因的方差,因此减少了下游分析的计算工作。 一个简单、高度有效和广泛使用的线性降维方法是主成分分析(PCA)。 最高的PCs将会捕捉数据集中占主导地位的异质性因子。
这里,我们将PCA用于缩放后的数据。 默认情况下,只有之前找到的1500个高度变化基因会被使用,并且前50个主成分会被计算和返回。 PCA结果可以通过PCA图来可视化。
N1469 <- RunPCA(N1469, features=VariableFeatures(N1469))
DimPlot(N1469, reduction = "pca")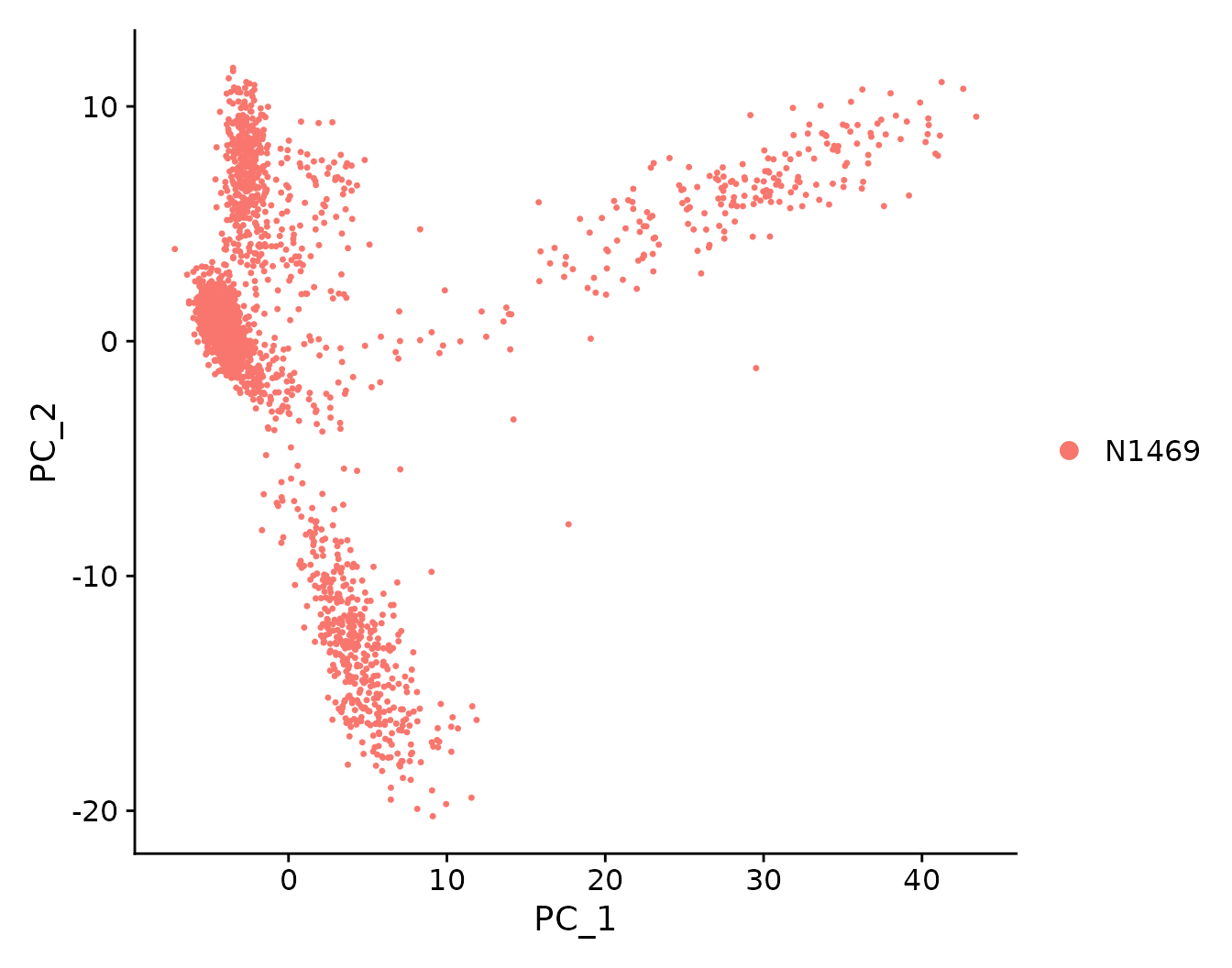
PCA图展示数据的前两个主成分
虽然PCA很大程度上减少了数据的维度,从几千个基因到50个PCs,但是同时可视化和解释50PCs依然很困难。 因此,为了更直观的理解数据,需要进一步的降维策略来将数据压缩到2-3维度。 两种流行的非线性降维技术是 t-stochastic neighbor embedding (tSNE) (Van der Maaten and Hinton 2008) 和 uniform manifold approximation and projection (UMAP) (McInnes, Healy, and Melville 2018)。
UMAP和tSNE可视化方法哪个更好是有争议。 UMAP倾向于具有更紧凑的可视化集群,但是减少了每个集群内的分辨率。 UMAP越来越少欢迎的主要原因是它比tSNE更快。 注意UMAP和tSNE都包括一些列随机步骤,所以设置seed是很关键的。
这里我们同时使用了UMAP和tSNE来进行数据降维和可视化。 前30个PC被用作输入数据,并且一个随机seed被使用以重现此结果。
dimUsed <- 30
N1469 <- RunUMAP(N1469, dims=1:dimUsed, seed.use=2021, verbose=FALSE)
N1469 <- RunTSNE(N1469, dims=1:dimUsed, seed.use=2021)
plot1 <- DimPlot(N1469, reduction = "umap")
plot2 <- DimPlot(N1469, reduction = "tsne")
plot1 + plot2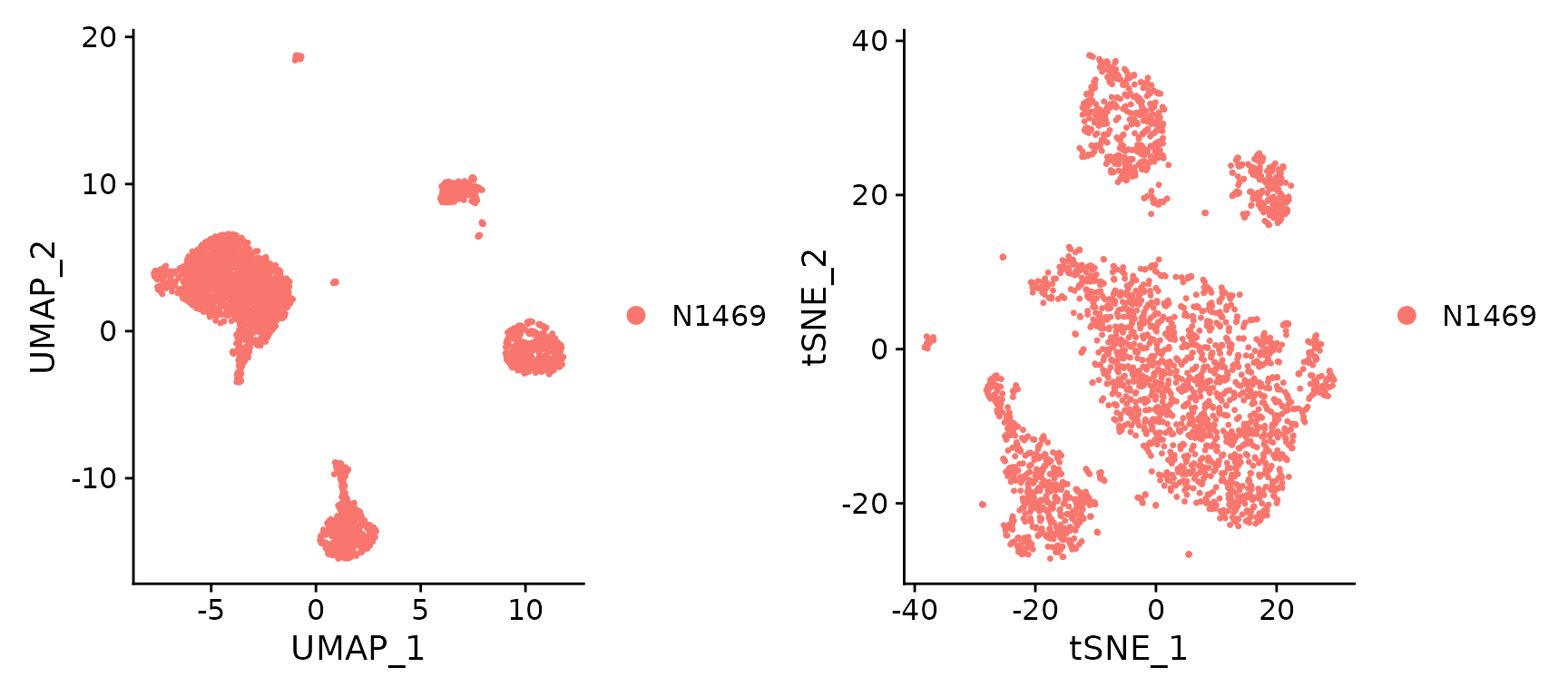
UMAP and t-SNE visualization
细胞聚类
细胞聚类是单细胞RNA-seq数据分析中将具有相似表达谱的细胞聚在一起的步骤。 这是总结数据的信息和提供数据的生物学解释的一个重要步骤。 Seurat提供了一个基于图的聚类方法,因其灵活性和可扩展性使它成为最流行的聚类方法之一。
在细胞聚类中经常会被问到的问题是:“在数据中应该有多少个cluster?” 这个问题经常很难回答,因为我们可以定义我们想要的cluster的数目。 实际上,cluster的数目将依赖于我们感兴趣的生物学问题(比如,主要细胞类型的分辨率是否足够或者亚型的分辨率是否需要)。 实践中,我们经常会尝试不同的分辨率来探究数据以获得“最佳”的分辨率来给出我们感兴趣问题的最佳解答。
在Seurat中,细胞聚类过程从构建一个KNN图开始,使用FindNeighbors()函数。 这里,我们用了前30PCs做为输入数据。 然后Seurat中的FindClusters()函数会用Louvain算法(默认)将细胞聚在一起。 对于这套特定数据,我们设置的分辨率参数是0.1。 最终的cluster可以通过Idents()函数找到。
N1469 <- FindNeighbors(N1469, dims=1:dimUsed)
N1469 <- FindClusters(N1469, resolution=0.1)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 2868
## Number of edges: 109080
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9600
## Number of communities: 5
## Elapsed time: 0 seconds##
## 0 1 2 3 4
## 1699 507 421 213 28我们也可以从metadata中获得最终的cluster信息。
colnames(N1469@meta.data)## [1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt"
## [5] "RNA_snn_res.0.1" "seurat_clusters"
table(N1469@meta.data$seurat_clusters)##
## 0 1 2 3 4
## 1699 507 421 213 28我们可以用UMAP图来可视化细胞clusters。
DimPlot(N1469, reduction = "umap", label = TRUE)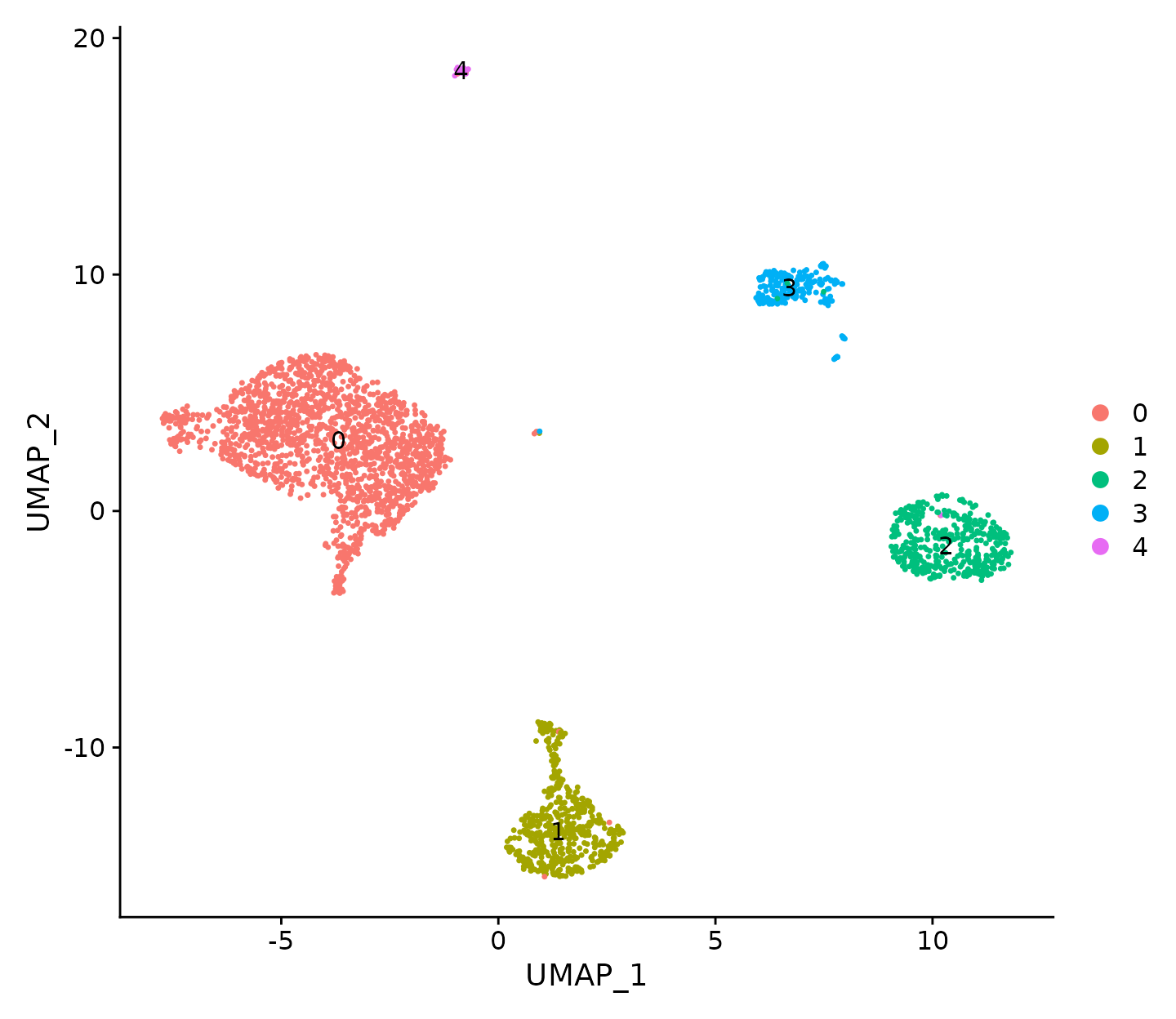
UMAP可视化图,细胞按细胞cluster着色
标记基因鉴定
细胞聚类的下一步是鉴定驱使细胞clusters分离的标记基因。 标记基因的获取通常是通过进行不同cluster之间的差异表达分析。 在Seurat中,差异表达分析由FindMarkers() 函数来完成。 默认情况下,它会比较单个cluster(由ident.1指定)和剩下的其他所有clusters,来鉴定该cluster的阳性和阴性标记基因。
为了增加计算速度,Seurat中的FindMarkers()函数只对一些满足特定阈值的基因子集进行DE检验。 参数min.pct要求基因在两组细胞中被检测到的百分比要超过一个阈值,这个阈值默认情况下是10%。 参数logfc.threshold限制被检测的基因在两组细胞中的log-fold change要高于一个特定水平(默认是0.25)。 默认情况下,FindMarkers() 函数会使用Wilcoxon Rank Sum检验,但是其他的统计检验方法(比如, likelihood-ratio检验, t-检验) 也是可以使用的。
这里我们找到了cluster 1的所有标记基因。
cluster1.markers <- FindMarkers(N1469, ident.1 = 1, min.pct = 0.25)
head(cluster1.markers)## p_val avg_log2FC pct.1 pct.2 p_val_adj
## CXCL13 0.00e+00 4.17 0.615 0.016 0.00e+00
## DNAJC12 0.00e+00 2.59 0.783 0.027 0.00e+00
## PTHLH 0.00e+00 2.87 0.698 0.016 0.00e+00
## BATF 0.00e+00 2.15 0.684 0.034 0.00e+00
## TFF3 0.00e+00 3.46 0.803 0.010 0.00e+00
## AGR2 3.05e-304 2.22 0.568 0.007 4.85e-300为了找到将cluster2区别于cluster 0和1的标记基因,我们可以用如下方法。
cluster2.markers <- FindMarkers(N1469, ident.1 = 2, ident.2 = c(0, 1), min.pct = 0.25)
head(cluster2.markers)## p_val avg_log2FC pct.1 pct.2 p_val_adj
## ACTG2 0.00e+00 5.09 0.789 0.035 0.0e+00
## CAV1 0.00e+00 3.55 0.891 0.092 0.0e+00
## TPM2 0.00e+00 3.63 0.900 0.068 0.0e+00
## KRT5 0.00e+00 3.79 0.929 0.138 0.0e+00
## KRT14 0.00e+00 5.40 0.969 0.057 0.0e+00
## SERPINB5 2.57e-292 2.26 0.686 0.032 4.1e-288Seurat中的FindAllMarkers()函数会自动化所有clusters的标记基因检测过程。 这里我们找到了每个cluster的标记基因,通过将该cluster的细胞与剩下所有细胞进行比较后得到,只报告阳性标记基因。
N1469.markers <- FindAllMarkers(N1469, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)我们选择每个cluster的最显著的3个标记基因,并将它们列在下方。
topMarkers <- split(N1469.markers, N1469.markers$cluster)
top3 <- lapply(topMarkers, head, n=3)
top3 <- do.call("rbind", top3)
top3## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
## 0.MMP7 0 4.06 0.957 0.216 0 0 MMP7
## 0.PDZK1IP1 0 2.50 0.950 0.314 0 0 PDZK1IP1
## 0.RPL12 0 1.29 0.999 0.974 0 0 RPL12
## 1.CXCL13 0 4.17 0.615 0.016 0 1 CXCL13
## 1.TFF3 0 3.46 0.803 0.010 0 1 TFF3
## 1.PTHLH 0 2.87 0.698 0.016 0 1 PTHLH
## 2.KRT14 0 5.41 0.969 0.056 0 2 KRT14
## 2.ACTG2 0 5.09 0.789 0.037 0 2 ACTG2
## 2.KRT5 0 3.80 0.929 0.134 0 2 KRT5
## 3.DCN 0 5.76 0.920 0.015 0 3 DCN
## 3.AKR1C1 0 4.97 0.859 0.024 0 3 AKR1C1
## 3.APOD 0 4.56 0.770 0.026 0 3 APOD
## 4.UBE2C 0 2.71 1.000 0.006 0 4 UBE2C
## 4.CDK1 0 1.48 0.679 0.001 0 4 CDK1
## 4.MKI67 0 1.37 0.714 0.000 0 4 MKI67最显著标记基因的表达水平可以被覆盖到UMAP图上来进行可视化。
FeaturePlot(N1469, features = top3$gene, ncol=3)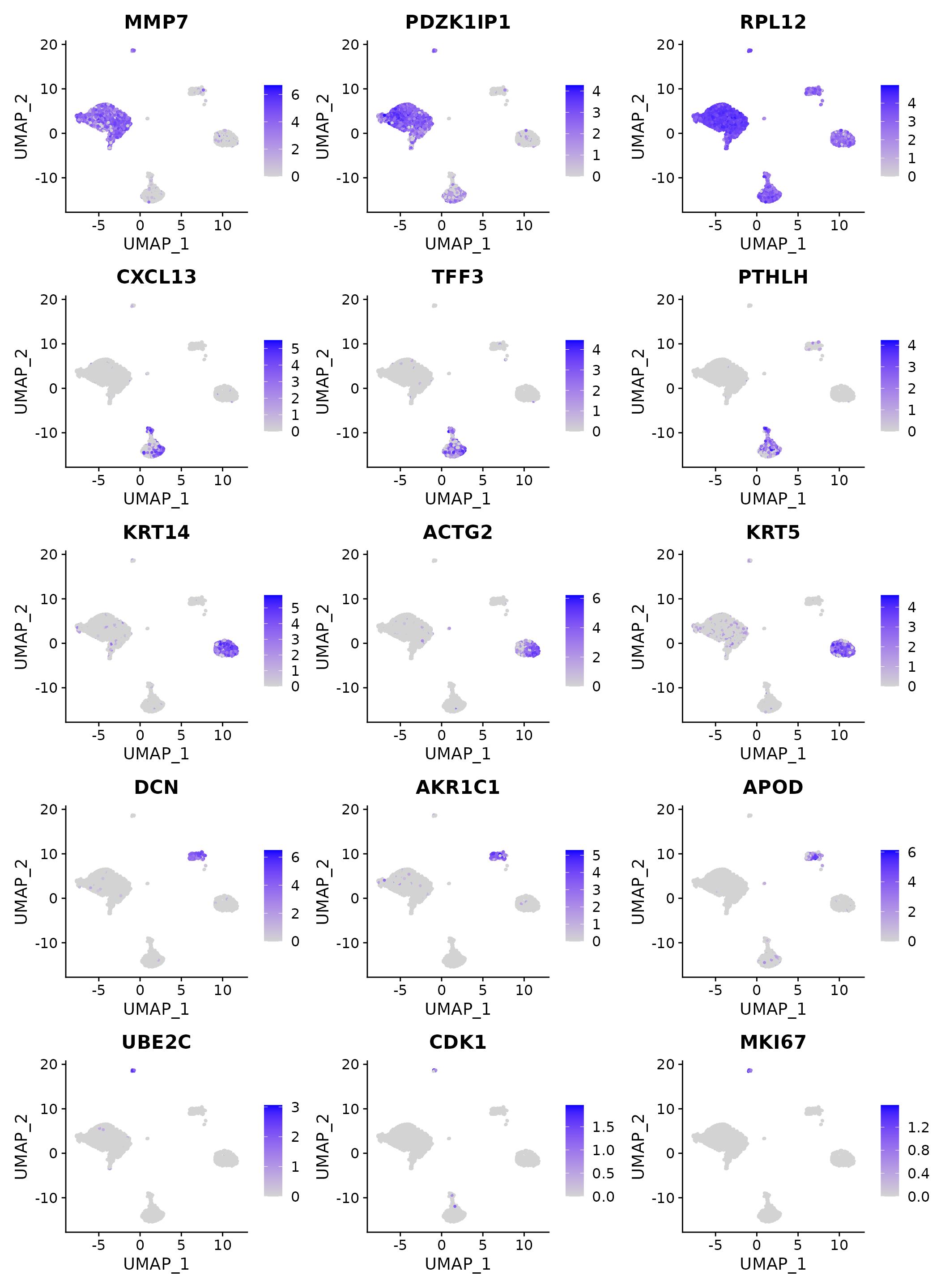
最显著标记基因表达的UMAP图可视化
点图也可以用来可视化最显著的标记基因。
DotPlot(N1469, features = top3$gene, dot.scale = 8) + RotatedAxis()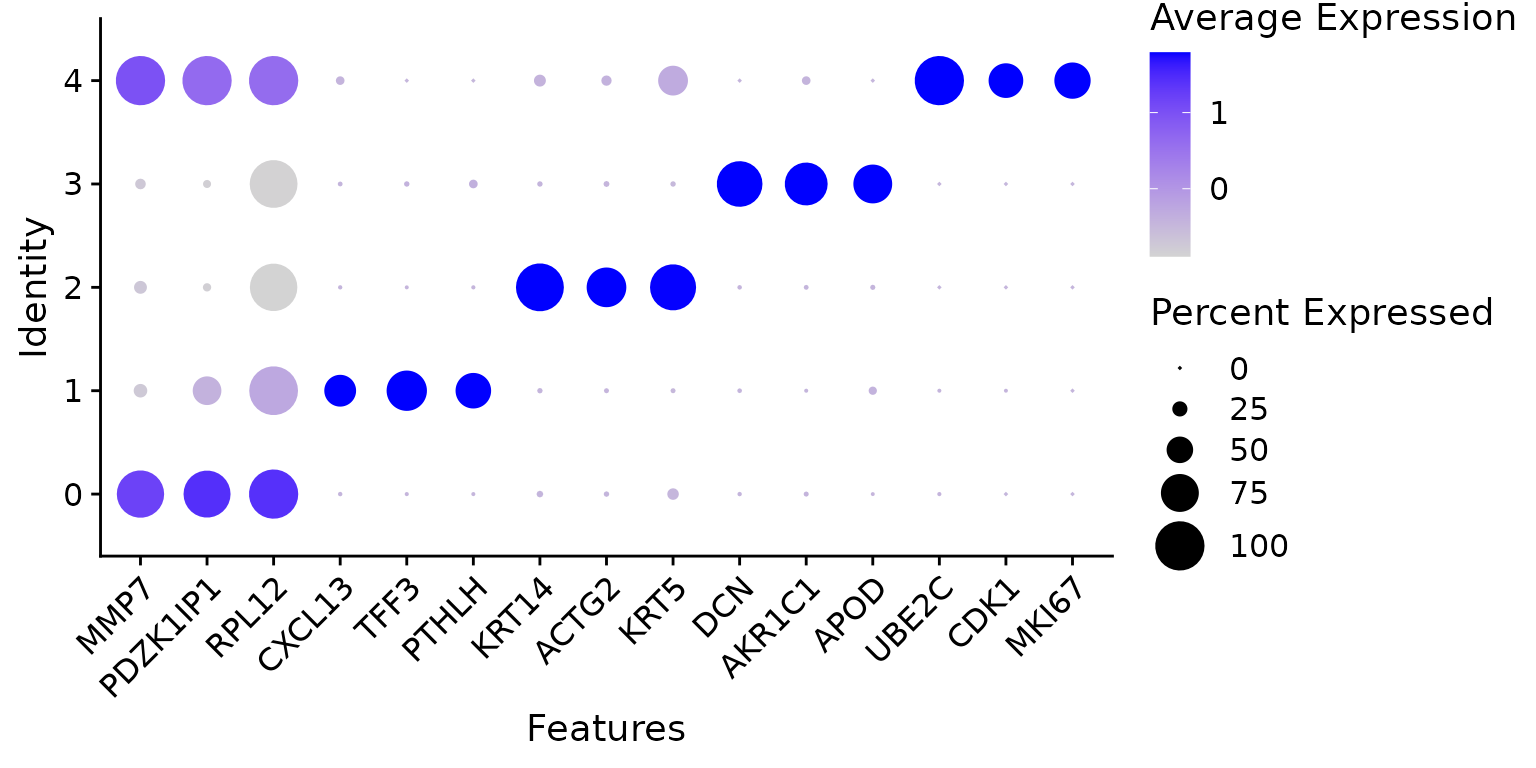
每个cluster的最显著基因的点图
细胞类型注释
在生物学背景下解释细胞clusters是scRNA-seq数据分析中最具有挑战性的任务之一。 通常需要先验生物学知识来做到这点。 来自于文献或curated基因集(比如,Gene Ontology, KEGG pathways)的标记基因是常见的先验信息来源。 或者,我们也可以用具有详细细胞注释信息的已发表的参考数据集。
这里我们使用了来自于同一研究 (Pal et al. 2021)的人类乳腺表皮的bulk RNA-seq数据集作为参考数据。 这套参考RNA-seq数据(GSE161892)总共包含来自basal,luminal progenitor (LP),mature luminal (ML)和stromal细胞类型的34个样本。 这套参考数据的差异表达分析使用的是limma-voom和TREAT (Law et al. 2014; McCarthy and Smyth 2009)。 如果基因在某种细胞类型中相对于其他所有细胞类型是上调的,那么这些基因被认为是细胞类型特异的。 这样产生了515个basal基因,323个LP基因,765个ML基因和1094个stroma基因。
这里我么下载了那些signature基因并将它们加载到R里面。
url.Signatures <- "https://github.com/SmythLab/scRNAseq-Workshop/raw/main/Data/Human-PosSigGenes.RData"
utils::download.file(url.Signatures, destfile="Data/Human-PosSigGenes.RData", mode="wb") 我们限制signature基因要在单细胞数据中表达。
load("Data/Human-PosSigGenes.RData")
HumanSig <- list(Basal=Basal, LP=LP, ML=ML, Str=Str)
HumanSig <- lapply(HumanSig, intersect, rownames(N1469))为了将单细胞数据中的每个细胞与参考bulk数据的四种细胞类群相关联,我们为每个细胞计算了这四种细胞类群的signature score。 每一种细胞类型的signature score被定义为一个细胞中这种细胞类型特定基因的平均表达水平。
SigScore <- list()
for(i in 1:length(HumanSig)){
SigScore[[i]] <- colMeans(N1469@assays$RNA@data[HumanSig[[i]], ])
}我们可以用UMAP图和小提琴图来可视化signature scores。
SigScores <- do.call("cbind", SigScore)
colnames(SigScores) <- c("Basal", "LP", "ML", "Stroma")
N1469@meta.data <- cbind(N1469@meta.data, SigScores)
FeaturePlot(N1469, features = colnames(SigScores))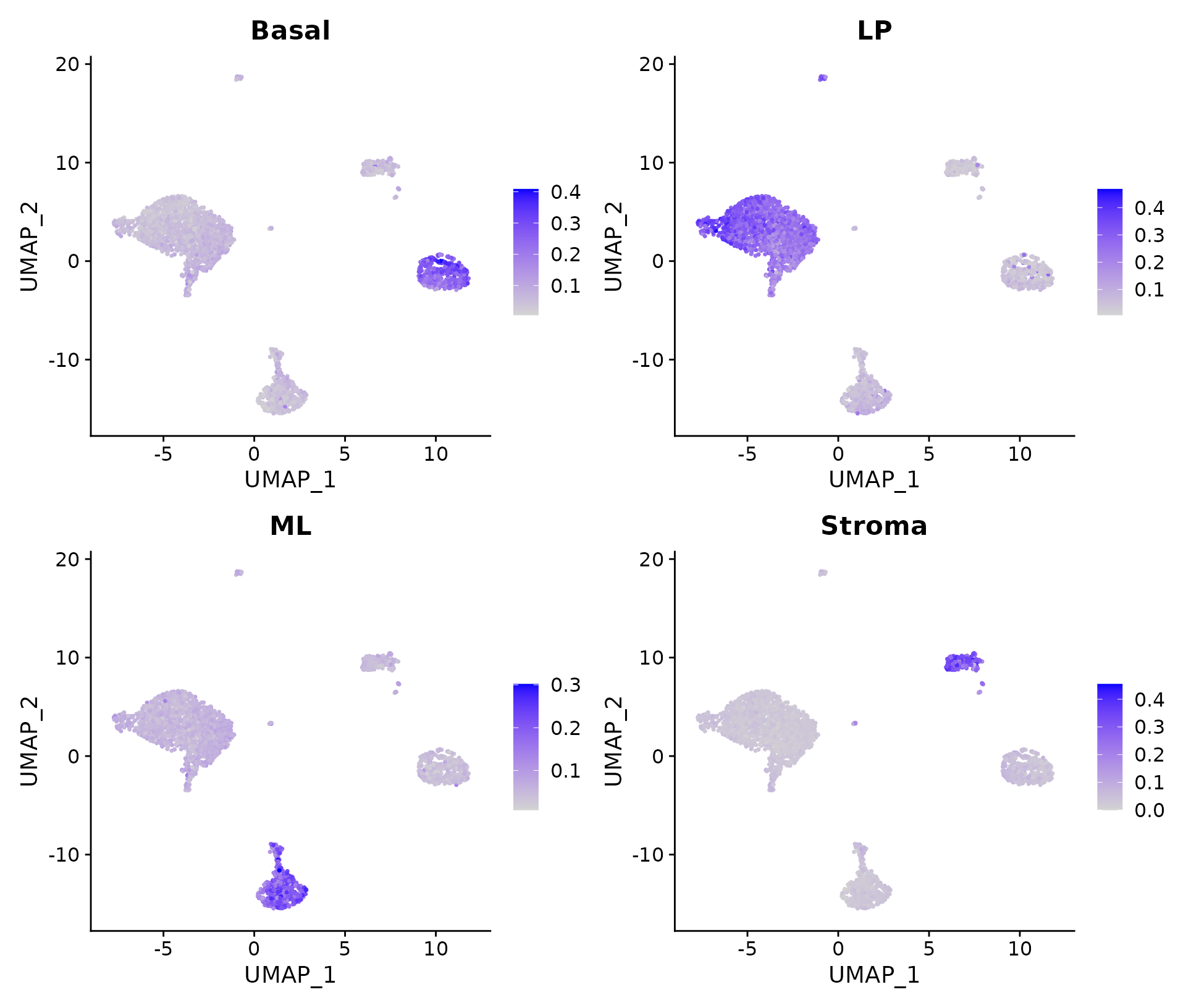
Signature scores
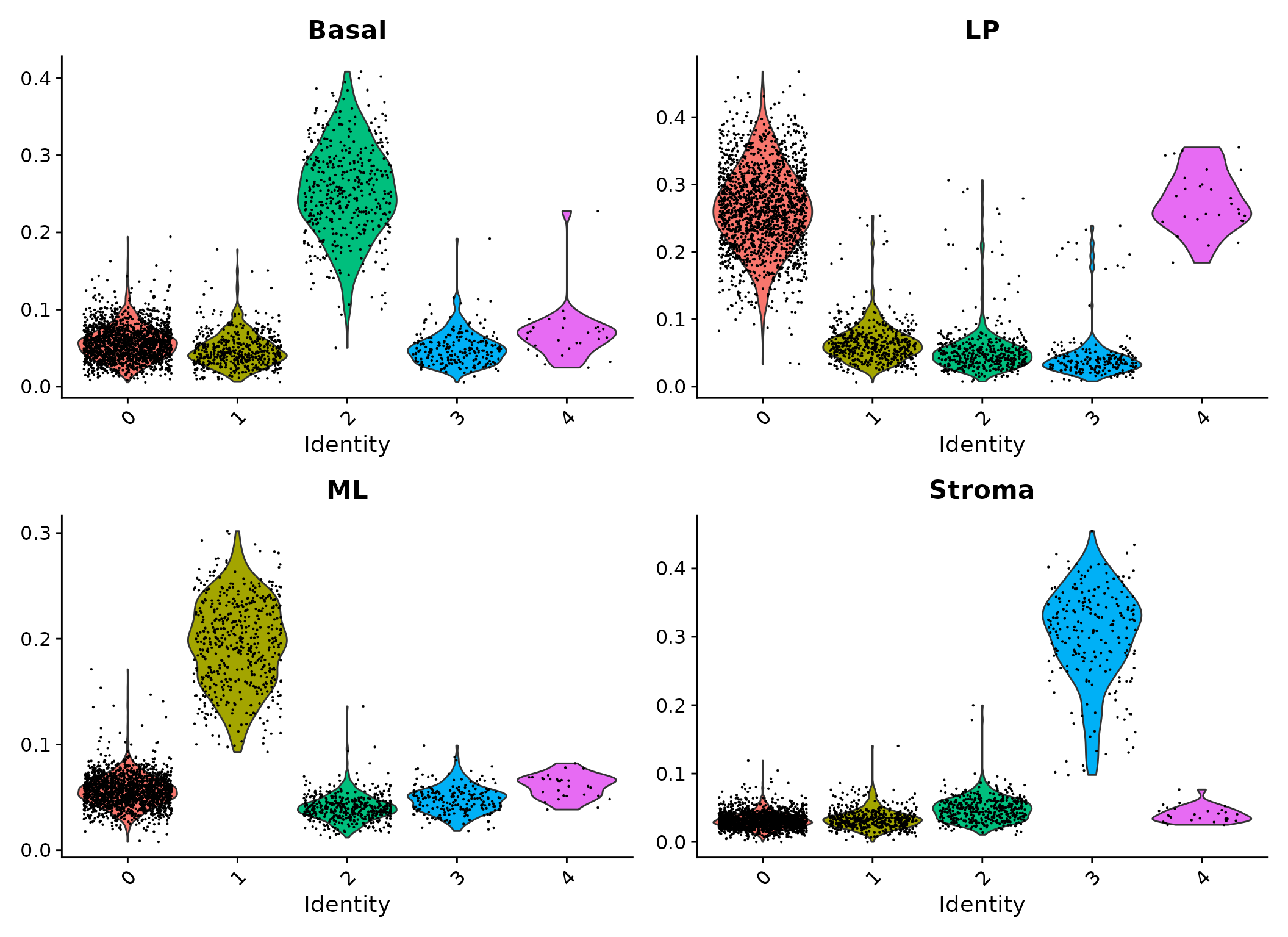
signature scores小提琴图
我们可以看到basal signature基因在cluster 2中具有较高表达水平,意味着这些细胞很可能是basal细胞。 相似的,cluster 0和4是LP,cluster 1是ML,cluster 3是stroma。
三元图分析
另一种可视化单细胞和参考bulk数据间相关性的方法是画三元图,这种方法在注重于三个主要细胞类群的研究者很有用。 对于这套数据,我们对如何将每个细胞分配给三种主要的表皮细胞类型(basal,LP和ML)之一很感兴趣。
我们生成了一个三元图来看细胞更接近于三种细胞类群中的哪一种。 为了评估每个细胞和三种类群之间的相似性,我们计数了每个细胞中表达的(至少有一个count)signature基因个数。 每个细胞在三元图上的位置由该细胞中表达的三种细胞类群的signature基因数目决定。
TN <- matrix(0L, ncol(N1469), 3L)
colnames(TN) <- c("LP", "ML", "Basal")
for(i in colnames(TN)){
TN[, i] <- colSums(N1469@assays$RNA@counts[HumanSig[[i]], ] > 0L)
}
head(TN)## LP ML Basal
## [1,] 46 34 16
## [2,] 39 21 12
## [3,] 49 42 24
## [4,] 42 33 11
## [5,] 14 106 21
## [6,] 38 26 17
library(vcd)
col.p <- scales::hue_pal()(nlevels(Idents(N1469)))
ternaryplot(TN, cex=0.2, pch=16, col=col.p[Idents(N1469)], grid=TRUE)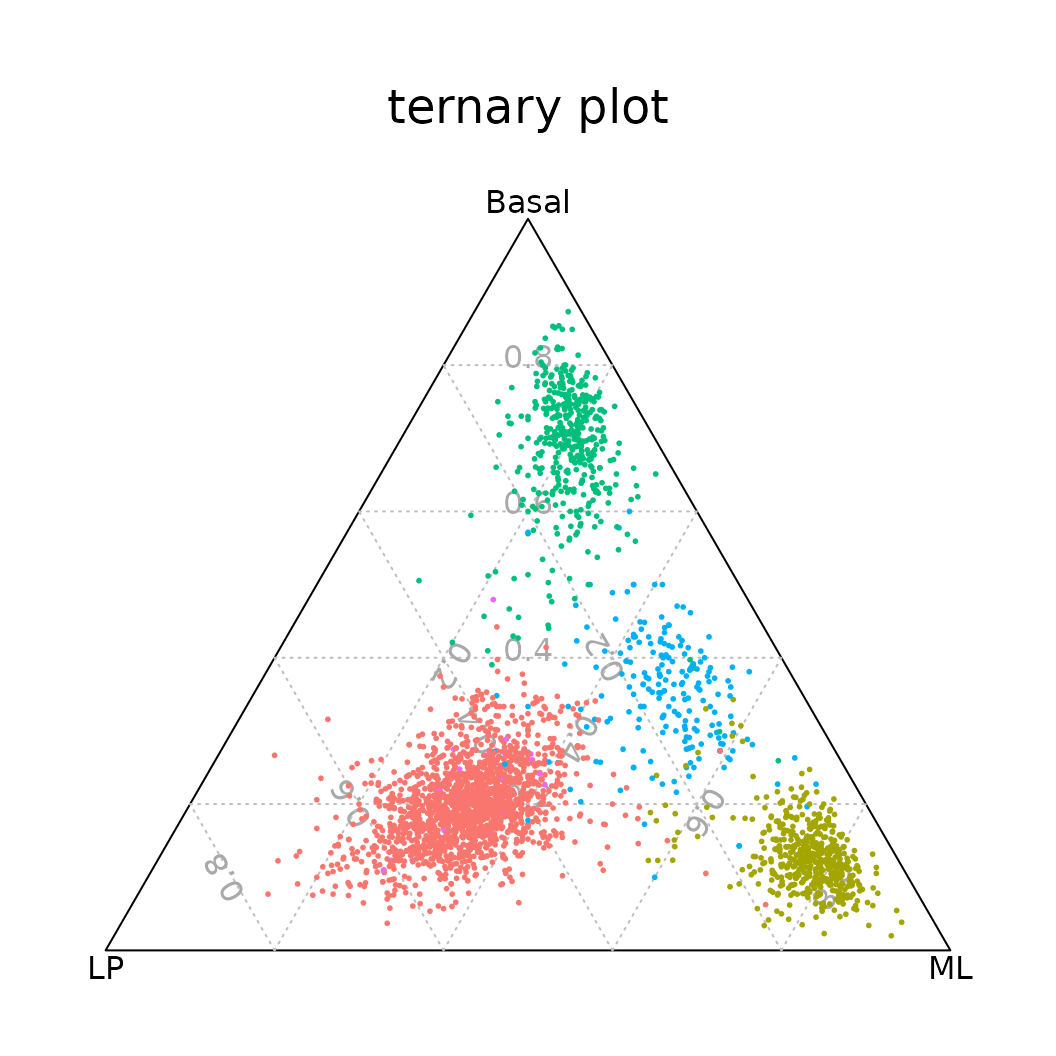
三元图,根据每个细胞表达的basal, LP, 和ML signature基因数目确定细胞位置
或者,用细胞表达的signature基因的相对比例来进行三元图分析。
## LP ML Basal
## 266 643 384## LP ML Basal
## [1,] 0.1729 0.0529 0.0417
## [2,] 0.1466 0.0327 0.0312
## [3,] 0.1842 0.0653 0.0625
## [4,] 0.1579 0.0513 0.0286
## [5,] 0.0526 0.1649 0.0547
## [6,] 0.1429 0.0404 0.0443
ternaryplot(TN_prop, cex=0.2, pch=16, col=col.p[Idents(N1469)], grid=TRUE)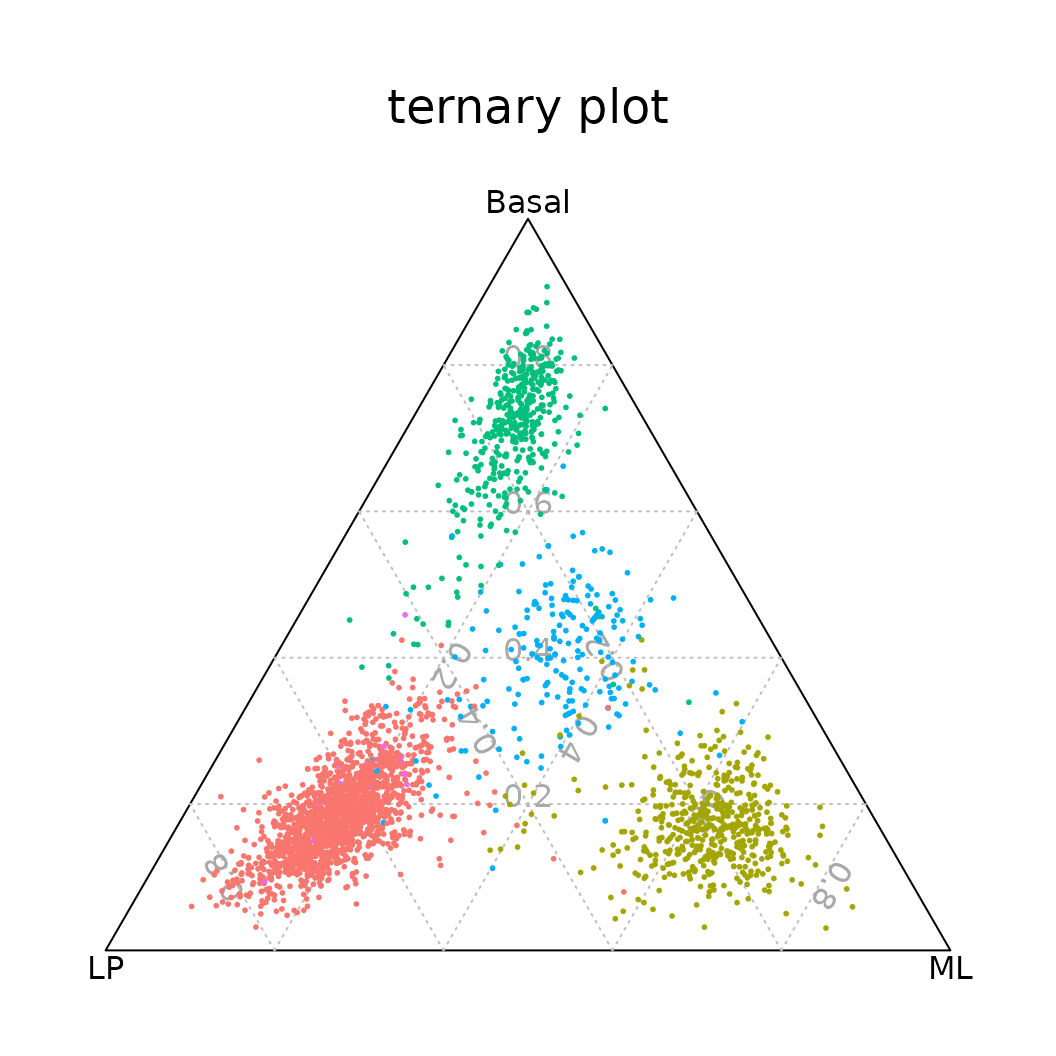
三元图,根据每个细胞表达的basal, LP, 和ML signature基因相对比例确定细胞位置
Session信息
## R version 4.1.1 (2021-08-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.3 LTS
##
## Matrix products: default
## BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
## [4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] vcd_1.4-8 SeuratObject_4.0.2 Seurat_4.0.4
##
## loaded via a namespace (and not attached):
## [1] Rtsne_0.15 colorspace_2.0-2 deldir_0.2-10
## [4] ellipsis_0.3.2 ggridges_0.5.3 rprojroot_2.0.2
## [7] fs_1.5.0 spatstat.data_2.1-0 farver_2.1.0
## [10] leiden_0.3.9 listenv_0.8.0 ggrepel_0.9.1
## [13] RSpectra_0.16-0 fansi_0.5.0 codetools_0.2-18
## [16] splines_4.1.1 cachem_1.0.6 knitr_1.34
## [19] polyclip_1.10-0 jsonlite_1.7.2 ica_1.0-2
## [22] cluster_2.1.2 png_0.1-7 uwot_0.1.10
## [25] shiny_1.7.0 sctransform_0.3.2 spatstat.sparse_2.0-0
## [28] compiler_4.1.1 httr_1.4.2 Matrix_1.3-4
## [31] fastmap_1.1.0 lazyeval_0.2.2 limma_3.49.4
## [34] later_1.3.0 htmltools_0.5.2 tools_4.1.1
## [37] igraph_1.2.6 gtable_0.3.0 glue_1.4.2
## [40] RANN_2.6.1 reshape2_1.4.4 dplyr_1.0.7
## [43] Rcpp_1.0.7 scattermore_0.7 jquerylib_0.1.4
## [46] pkgdown_1.6.1 vctrs_0.3.8 nlme_3.1-153
## [49] lmtest_0.9-38 xfun_0.26 stringr_1.4.0
## [52] globals_0.14.0 mime_0.11 miniUI_0.1.1.1
## [55] lifecycle_1.0.1 irlba_2.3.3 goftest_1.2-2
## [58] future_1.22.1 MASS_7.3-54 zoo_1.8-9
## [61] scales_1.1.1 spatstat.core_2.3-0 ragg_1.1.3
## [64] promises_1.2.0.1 spatstat.utils_2.2-0 parallel_4.1.1
## [67] RColorBrewer_1.1-2 yaml_2.2.1 memoise_2.0.0
## [70] reticulate_1.22 pbapply_1.5-0 gridExtra_2.3
## [73] ggplot2_3.3.5 sass_0.4.0 rpart_4.1-15
## [76] stringi_1.7.4 highr_0.9 desc_1.3.0
## [79] rlang_0.4.11 pkgconfig_2.0.3 systemfonts_1.0.2
## [82] matrixStats_0.61.0 evaluate_0.14 lattice_0.20-45
## [85] ROCR_1.0-11 purrr_0.3.4 tensor_1.5
## [88] labeling_0.4.2 patchwork_1.1.1 htmlwidgets_1.5.4
## [91] cowplot_1.1.1 tidyselect_1.1.1 parallelly_1.28.1
## [94] RcppAnnoy_0.0.19 plyr_1.8.6 magrittr_2.0.1
## [97] R6_2.5.1 generics_0.1.0 withr_2.4.2
## [100] mgcv_1.8-37 pillar_1.6.2 fitdistrplus_1.1-5
## [103] survival_3.2-13 abind_1.4-5 tibble_3.1.4
## [106] future.apply_1.8.1 crayon_1.4.1 KernSmooth_2.23-20
## [109] utf8_1.2.2 spatstat.geom_2.2-2 plotly_4.9.4.1
## [112] rmarkdown_2.11 data.table_1.14.0 digest_0.6.28
## [115] xtable_1.8-4 tidyr_1.1.3 httpuv_1.6.3
## [118] textshaping_0.3.5 munsell_0.5.0 viridisLite_0.4.0
## [121] bslib_0.3.0